DMREF Publications
Efficient Polymorph Screening through Crystallization from Bulk and Confined Melts

Crystallization from the melt can allow the achievement of high driving force for crystallization accompanied by relatively slow growth, nucleation, and transformation rates, features that favor its use as an efficient polymorph screening method. Surprisingly, even though melt crystallization has a long history, it has been employed less often in the search for new polymorphs than solution crystallization. Applications of melt crystallization to 21 highly polymorphic, well-characterized compounds with at least five ambient polymorphs revealed that melt crystallization afforded more than half of the known polymorphs and in many cases revealed new polymorphs not detected by other screening methods. A statistical analysis revealed that polymorphs grown from the melt have a greater propensity for high Z′ values, which are not easily accessible by other crystallization protocols and are often not detectable by crystal structure prediction methods. Melt crystallization within nanopores (8−100 nm) performed for 19 of the 21 compounds mostly resulted in polymorphs that dominated crystallization from the bulk melt at similar temperatures. The total number of polymorphs observed in nanopores was less than that observed during crystallization from the bulk melt, however, and melt crystallization under confinement revealed new polymorphs not detected by other crystallization methods.
Read the article here.
N. Fellah, L. Tahsin, C. Jin Zhang, B. Kahr, M. D. Ward, A. G. Shtukenberg, Cryst. Growth Des. 2022, 22, 7527–7543.
Geometric Deep Learning for Molecular Crystal Structure Prediction

We develop and test new machine learning strategies for accelerating molecular crystal structure ranking and crystal property prediction using tools from geometric deep learning on molecular graphs. Leveraging developments in graph-based learning and the availability of large molecular crystal data sets, we train models for density prediction and stability ranking which are accurate, fast to evaluate, and applicable to molecules of widely varying size and composition. Our density prediction model, MolXtalNet-D, achieves state-of-the-art performance, with lower than 2% mean absolute error on a large and diverse test data set. Our crystal ranking tool, MolXtalNet-S, correctly discriminates experimental samples from synthetically generated fakes and is further validated through analysis of the submissions to the Cambridge Structural Database Blind Tests 5 and 6. Our new tools are computationally cheap and flexible enough to be deployed within an existing crystal structure prediction pipeline both to reduce the search space and score/filter crystal structure candidates.
Read the article here
M. Kilgour, J. Rogal, M. Tuckerman, J. Chem. Theory Comput. 2023, 19, 4743–4756.
Chlorfenapyr Crystal Polymorphism and Insecticidal Activity

Optical micrographs of chlorfenapyr polymorphs. Polarizers are crossed for images (A–E). (A) Form I was crystallized from the melt at 80 °C. (B) Form I crystallized from the melt at 23 °C. (C) Transformation of form I into form II/III starting from the edge of the slide at the lower left corner. (D) Feathery flakes of form IV grown from the melt and surrounded by spherulites of form I. (E) Spherulite form IV nucleated at 50 °C. (F) Crystal of form III grown from hexanes. (G) Needle of form II formed on the surface of a poly(ethylene) fiber. If not specified, the scale bars are 0.5 mm..
The search for new insecticides - a.k.a. the “insecticide treadmill - with new mechanisms of action for the control of rapidly reproducing organisms that transmit disease continues. Malarial (Anopheles) mosquitoes are now widely resistant to pyrethroids, synthetic compounds related to pyrethrum, a natural insecticide found in chrysanthemums. Since the 1980s, pyrethroids have shouldered the lion’s share of malaria control. For decades, no new adulticides for vector control were approved by the World Health Organization (WHO). In the face of worldwide pyrethroid resistance, however, the WHO approved chlorfenapyr, a pyrrole proinsecticide typically used alongside pyrethroids to combat pyrethroid resistance. Here, DMREF investigators discovered four crystalline polymorphs of chlorfenapyr, three of which are. considered polytypic. Chlorfenapyr polymorphs show similar lethality against fruit flies (Drosophila melanogaster) and mosquitoes (Anopheles quadrimaculatus) with the least stable polymorph showing slightly higher lethality. Knockdown kinetics, however, depend on an internal metabolic activating step, which further complicates polymorph-dependent bioavailability.
Read the article here.
R. Aronin, P. Brázda, L. N. Smith, C. J. Zhang, J. B. Benedict, Z. Y. Marr, B. Rybtchinski, H. Weissman, A. G. Shtukenberg, B. Kahr, Cryst. Growth Des. 2024, XXXX, XXX, XXX-XXX (https://doi.org/10.1021/acs.cgd.3c01257)
Topological Crystal Structure Prediction

Organic molecular crystals constitute a class of materials of critical importance in numerous industries. Despite the ubiquity of these systems, our ability to predict molecular crystal structures starting only from a two-dimensional diagram of the constituent compound(s) remains a significant challenge. Most structure-prediction protocols require a customized interatomic interaction model on which the quality of the results can depend sensitively. To overcome this problem, we introduce a new topological approach to molecular crystal structure prediction. The approach posits that in a stable structure, molecules are oriented such that principal axes and normal ring plane vectors are aligned with specific crystallographic directions and that heavy atoms occupy positions that correspond to minima of a set of geometric order parameters. By minimizing an objective function that encodes these orientations and atomic positions, and filtering based on the vdW free volume and intermolecular close contacts distributions derived from the Cambridge Structural Database, stable structures and polymorphs for a given crystal can be predicted entirely mathematically without reliance on an interaction model.
Read the article here.
N. Galanakis, M. Tuckerman, Nat. Commun. 2024, 15, 9757
Suppressing Disorder in Benzamide and Thiobenzamide Crystals by Fluorine Substitution
Disorder is a common feature of molecular crystals that complicates determination of structures and can potentially affect electric and mechanical properties. Suppression of disorder is observed in otherwise severely disordered benzamide and thiobenzamide crystals by substituting hydrogen with fluorine in the ortho-position of the phenyl ring. Fluorine occupancies of 20−30% are sufficient to suppress disorder without changing the packing motif. Crystal structure prediction calculations reveal a much denser lattice energy landscape for benzamide compared to 2-fluorobenzamide, suggesting that fluorine substitution makes disorder less likely.
Read the article here
A. G. Shtukenberg, D. E. Braun, M. Tan, N. Fellah, B. Kahr, Cryst. Growth Des. 2024, 24, 5276–5284.
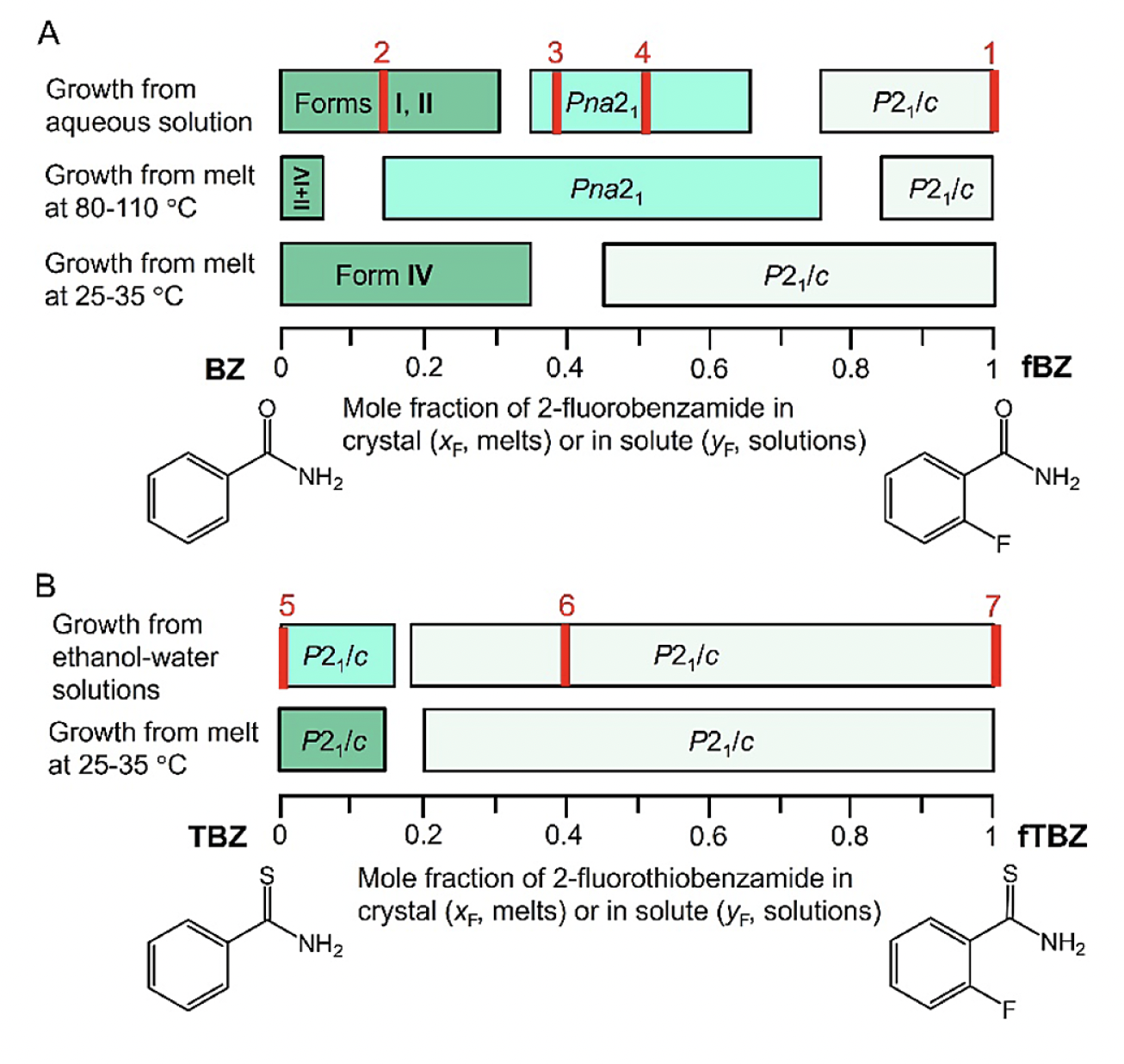
Polymorphism in benzamide − 2-fluorobenzamide (A) and thiobenzamide − 2-fluorothiobenzamide series (B). Increasing color intensity corresponds to increasing disorder. Red bars indicate compositions for which single crystal structure determinations have been realized.
Machine Learning Classification of Local Environments in Molecular Crystals
Identifying local structural motifs and packing patterns of molecular solids is a challenging task for both simulation and experiment. We demonstrate two novel approaches to characterize local environments in different polymorphs of molecular crystals using learning models that employ either flexibly learned or handcrafted molecular representations. In the first case, we follow our earlier work on graph learning in molecular crystals, deploying an atomistic graph convolutional network combined with molecule-wise aggregation to enable per-molecule environmental classification. For the second model, we develop a new set of descriptors based on symmetry functions combined with a point-vector representation of the molecules, encoding information about the positions and relative orientations of the molecule. We demonstrate very high classification accuracy for both approaches on urea and nicotinamide crystal polymorphs and practical applications to the analysis of dynamical trajectory data for nanocrystals and solid–solid interfaces. Both architectures are applicable to a wide range of molecules and diverse topologies, providing an essential step in the exploration of complex condensed matter phenomena.
Read the article here
D. Kuroshima, M. Kilgour, M. E. Tuckerman, J. Rogal, J. Chem. Theory Comput. 2024, 20, 6197−6206.
—————
New Publications (2025; more details and links to follow soon):
(1) Kilgour, Michael, Mark Tuckerman, and Jutta Rogal. "Multi-Type Point Cloud Autoencoder: A Complete Equivariant Embedding for Molecule Conformation and Pose." arXiv preprint arXiv:2405.13791 (2024).
Abstract:
The point cloud is a flexible representation for a wide variety of data types, and is a particularly natural fit for the 3D conformations of molecules. Extant molecule embedding/representation schemes typically focus on internal degrees of freedom, ignoring the global 3D orientation. For tasks that depend on knowledge of both molecular conformation and 3D orientation, such as the generation of molecular dimers, clusters, or condensed phases, we require a representation which is provably complete in the types and positions of atomic nuclei and roto-inversion equivariant with respect to the input point cloud. We develop, train, and evaluate a new type of autoencoder, molecular O(3) encoding net (Mo3ENet), for multi-type point clouds, for which we propose a new reconstruction loss, capitalizing on a Gaussian mixture representation of the input and output point clouds. Mo3ENet is end-to-end equivariant, meaning the learned representation can be manipulated on O(3), a practical bonus for downstream learning tasks. An appropriately trained Mo3ENet latent space comprises a universal embedding for scalar and vector molecule property prediction tasks, as well as other downstream tasks incorporating the 3D molecular pose.
(2) "Polymorph sampling with coupling to extended variables: enhanced sampling of polymorph energy landscapes and free energy perturbation of polymorph ensembles"
E. J. Chan and M. E. Tuckerman Acta Crystallographica Section B, volume 80, 575 (2024).
Abstract:
A novel approach to computationally enhance the sampling of molecular crystal structures is proposed and tested. This method is based on the use of extended variables coupled to a Monte Carlo based crystal polymorph generator. Inspired by the established technique of quasi-random sampling of polymorphs using the rigid molecule constraint, this approach represents molecular clusters as extended variables within a thermal reservoir. Polymorph unit-cell variables are generated using pseudo-random sampling. Within this framework, a harmonic coupling between the extended variables and polymorph configurations is established. The extended variables remain fixed during the inner loop dedicated to polymorph sampling, enforcing a stepwise propagation of the extended variables to maintain system exploration. The final processing step results in a polymorph energy landscape, where the raw structures sampled to create the extended variable trajectory are re-optimized without the thermal coupling term. The foundational principles of this approach are described and its effectiveness using both a Metropolis Monte Carlo type algorithm and modifications that incorporate replica exchange is demonstrated. A comparison is provided with pseudo-random sampling of polymorphs for the molecule coumarin. The choice to test a design of this algorithm as relevant for enhanced sampling of crystal structures was due to the obvious relation between molecular structure variables and corresponding crystal polymorphs as representative of the inherent vapor to crystal transitions that exist in nature. Additionally, it is shown that the trajectories of extended variables can be harnessed to extract fluctuation properties that can lead to valuable insights. A novel thermodynamic variable is introduced: the free energy difference between ensembles of Z ' = 1 and Z ' = 2 crystal polymorphs.
(3) "The seventh blind test of crystal structure prediction: structure generation methods"
Hunnisett, LM; Nyman, J; Francia, N; Abraham, NS; Adjiman, CS; Aitipamula, S; Alkhidir, T; Almehairbi, M; Anelli, A; Anstine, DM; Anthony, JE; Arnold, JE; Bahrami, F; Bellucci, MA; Bhardwaj, RM; Bier, I; Bis, JA; Boese, AD; Bowskill, DH; Bramley, J; Brandenburg, JG; Braun, DE; Butler, PWV; Cadden, J; Carino, S; Chan, EJ; Chang, C; Cheng, BQ; Clarke, SM; Coles, SJ; Cooper, RI; Couch, R; Cuadrado, R; Darden, T; Day, GM; Dietrich, H; Ding, YM; DiPasquale, A; Dhokale, B; van Eijck, BP; Elsegood, MRJ; Firaha, D; Fu, WB; Fukuzawa, K; Glover, J; Goto, H; Greenwell, C; Guo, R; Harter, J; Helfferich, J; Hofmann, DWM; Hoja, J; Hone, J; Hong, R; Hutchison, G; Ikabata, Y; Isayev, O; Ishaque, O; Jain, V; Jin, YD; Jing, AL; Johnson, ER; Jones, I; Jose, KVJ; Kabova, EA; Keates, A; Kelly, PF; Khakimov, D; Konstantinopoulos, S; Kuleshova, LN; Li, H; Lin, XL; List, A; Liu, CC; Liu, YM; Liu, ZH; Liu, ZP; Lubach, JW; Marom, N; Maryewski, AA; Matsui, H; Mattei, A; Mayo, RA; Melkumov, JW; Mohamed, S; Abardeh, ZM; Muddana, HS; Nakayama, N; Nayal, KS; Neumann, MA; Nikhar, R; Obata, S; O'Connor, D; Oganov, AR; Okuwaki, K; Otero-de-la-Roza, A; Pantelides, CC; Parkin,S; Pickard, CJ; Pilia, L; Pivina, T; Podeszwa, R; Price, AJA; Price, LS; Price, SL; Probert, MR; Pulido, A; Ramteke, GR; Rehman, AU; Reutzel-Edens, SM; Rogal, J; Ross, MJ; Rumson, AF; Sadiq, G; Saeed, ZM; Salimi, A; Salvalaglio, M; de Almada, LS; Sasikumar, K; Sekharan, S; Shang, C; Shankland, K; Shinohara, K; Shi, BM; Shi, XK; Skillman, AG; Song, HX; Strasser, N; van de Streek, J; Sugden, IJ; Sun, GX; Szalewicz, K; Tan, BI; Tan, L; Tarczynski, F; Taylor, CR; Tkatchenko, A; Tom, R; Tuckerman, ME; Utsumi, Y; Vogt-Maranto, L; Weatherston, J; Wilkinson, LJ; Willacy, RD; Wojtas, L; Woollam, GR; Yang, ZC; Yonemochi, E; Yue, X; Zeng, Q; Zhang, YZ; Zhou, T; Zhou, YF; Zubatyuk, R; Cole, JC Acta Cryst. B 80, 517 (2024).
Abstract:
A seventh blind test of crystal structure prediction was organized by the Cambridge Crystallographic Data Centre featuring seven target systems of varying complexity: a silicon and iodine-containing molecule, a copper coordination complex, a near-rigid molecule, a cocrystal, a polymorphic small agrochemical, a highly flexible polymorphic drug candidate, and a polymorphic morpholine salt. In this first of two parts focusing on structure generation methods, many crystal structure prediction (CSP) methods performed well for the small but flexible agrochemical compound, successfully reproducing the experimentally observed crystal structures, while few groups were successful for the systems of higher complexity. A powder X-ray diffraction (PXRD) assisted exercise demonstrated the use of CSP in successfully determining a crystal structure from a low-quality PXRD pattern. The use of CSP in the prediction of likely cocrystal stoichiometry was also explored, demonstrating multiple possible approaches. Crystallographic disorder emerged as an important theme throughout the test as both a challenge for analysis and a major achievement where two groups blindly predicted the existence of disorder for the first time. Additionally, large-scale comparisons of the sets of predicted crystal structures also showed that some methods yield sets that largely contain the same crystal structures.
(4) "The seventh blind test of crystal structure prediction: structure ranking methods"
Hunnisett, LM; Nyman, J; Francia, N; Abraham, NS; Adjiman, CS; Aitipamula, S; Alkhidir, T; Almehairbi, M; Anelli, A; Anstine, DM; Anthony, JE; Arnold, JE; Bahrami, F; Bellucci, MA; Bhardwaj, RM; Bier, I; Bis, JA; Boese, AD; Bowskill, DH; Bramley, J; Brandenburg, JG; Braun, DE; Butler, PWV; Cadden, J; Carino, S; Chan, EJ; Chang, C; Cheng, BQ; Clarke, SM; Coles, SJ; Cooper, RI; Couch, R; Cuadrado, R; Darden, T; Day, GM; Dietrich, H; Ding, YM; DiPasquale, A; Dhokale, B; van Eijck, BP; Elsegood, MRJ; Firaha, D; Fu, WB; Fukuzawa, K; Glover, J; Goto, H; Greenwell, C; Guo, R; Harter, J; Helfferich, J; Hofmann, DWM; Hoja, J; Hone, J; Hong, R; Hutchison, Galanakis, N; G; Ikabata, Y; Isayev, O; Ishaque, O; Jain, V; Jin, YD; Jing, AL; Johnson, ER; Jones, I; Jose, KVJ; Kabova, EA; Keates, A; Kelly, PF; Khakimov, D; Konstantinopoulos, S; Kuleshova, LN; Li, H; Lin, XL; List, A; Liu, CC; Liu, YM; Liu, ZH; Liu, ZP; Lubach, JW; Marom, N; Maryewski, AA; Matsui, H; Mattei, A; Mayo, RA; Melkumov, JW; Mohamed, S; Abardeh, ZM; Muddana, HS; Nakayama, N; Nayal, KS; Neumann, MA; Nikhar, R; Obata, S; O'Connor, D; Oganov, AR; Okuwaki, K; Otero-de-la-Roza, A; Pantelides, CC; Parkin,S; Pickard, CJ; Pilia, L; Pivina, T; Podeszwa, R; Price, AJA; Price, LS; Price, SL; Probert, MR; Pulido, A; Ramteke, GR; Rehman, AU; Reutzel-Edens, SM; Rogal, J; Ross, MJ; Rumson, AF; Sadiq, G; Saeed, ZM; Salimi, A; Salvalaglio, M; de Almada, LS; Sasikumar, K; Sekharan, S; Shang, C; Shankland, K; Shinohara, K; Shi, BM; Shi, XK; Skillman, AG; Song, HX; Strasser, N; van de Streek, J; Sugden, IJ; Sun, GX; Szalewicz, K; Tan, BI; Tan, L; Tarczynski, F; Taylor, CR; Tkatchenko, A; Tom, R; Tuckerman, ME; Utsumi, Y; Vogt-Maranto, L; Weatherston, J; Wilkinson, LJ; Willacy, RD; Wojtas, L; Woollam, GR; Yang, ZC; Yonemochi, E; Yue, X; Zeng, Q; Zhang, YZ; Zhou, T; Zhou, YF; Zubatyuk, R; Cole, JC Acta Cryst. B 80, 548 (2024).
Abstract:
A seventh blind test of crystal structure prediction has been organized by the Cambridge Crystallographic Data Centre. The results are presented in two parts, with this second part focusing on methods for ranking crystal structures in order of stability. The exercise involved standardized sets of structures seeded from a range of structure generation methods. Participants from 22 groups applied several periodic DFT-D methods, machine learned potentials, force fields derived from empirical data or quantum chemical calculations, and various combinations of the above. In addition, one non-energy-based scoring function was used. Results showed that periodic DFT-D methods overall agreed with experimental data within expected error margins, while one machine learned model, applying system-specific AIMnet potentials, agreed with experiment in many cases demonstrating promise as an efficient alternative to DFT-based methods. For target XXXII, a consensus was reached across periodic DFT methods, with consistently high predicted energies of experimental forms relative to the global minimum (above 4 kJ mol(-1) at both low and ambient temperatures) suggesting a more stable polymorph is likely not yet observed. The calculation of free energies at ambient temperatures offered improvement of predictions only in some cases (for targets XXVII and XXXI). Several avenues for future research have been suggested, highlighting the need for greater efficiency considering the vast amounts of resources utilized in many cases.
(5) Polymer-Assisted Polymorph Transition in Melt-Processed Molecular Semiconductor Crystals
Pallavi Sundaram,∥ Rochelle B. Spencer,∥ Akash Tiwari, St. John Whittaker, Trinanjana Mandal, Yongfan Yang, Emma K. Holland, Christopher J. Kingsbury, Mia Klopfenstein, John E. Anthony, Bart Kahr, Sehee Jeong,* Alexander G. Shtukenberg,* and Stephanie S. Lee*
Chem. Mater. 2024, 36, 5976−5985
ABSTRACT:
A previously unreported polymorph of 5,11-bis-(triisopropylsilylethynyl)anthradithiophene (TIPS ADT), Form II, crystallizes from melt-processed TIPS ADT films blended with 16 ± 1 wt % medium density polyethylene (PE). TIPS ADT/PE blends that initially are crystallized from the melt produce twisted TIPS ADT crystals of a metastable polymorph (Form IV, space group P1̅) with a brickwork packing motif distinct from the slipstack packing by solution-processed TIPS ADT crystals (Form I, space group P21/c) at room temperature. When these films were cooled to room temperature and subsequently annealed at 100 °C, near a PE melting temperature of 110 °C, Form II crystals nucleated and grew while consuming Form IV. The growth rate and orientations of Form II crystals were predetermined by the twisting pitch and growth direction of the original banded spherulites in the melt-processed films of the blends. Notably, the Form IV → II transition was not observed during thermal annealing of neat TIPS ADT films without PE. The presence of the mobile PE phase during thermal annealing of TIPS ADT/PE blend films increases the diffusion rate of TIPS ADT molecules, and the rate of nucleation of Form II. Form IV crystals are more conductive but less emissive compared to Form II crystals.
